Heritable Platelet Disorders (BSH 2021)
N.B. There are several useful case studies at the end of BSH guideline
Normal Platelet Function

clinical assessment
Numerical & functional platelet disorders can co-exist with other bleeding disorders and be indistinguishable from one another.
Symptoms include easy bruising, prolonged bleeding, epistaxis, gum bleeding, GI/GU bleeding, post-procedure bleeding. In children ask about cephalohaematoma, umbilical cord bleeding, heel prick beeding.
(Joint / Muscle / Brain bleeding less common in HPD’s compared to haemophilia)
Can be useful to quantify with a bleeding assessment tool (e.g. ISTH-BAT) but be aware this was validated for VWD not platelet disorders.
Examination should look for phenotypic evidence of recognised syndromes (e.g. TAR) and an assessment for hypermobility (Beighton score)
Diagnosis should be by consensus of a MDT review of the clinical and laboratory features
testing - general considerations
Testing of platelet function highly sensitive to pre-analytical variables
Collect sample from a fasted, rested subject
Large bore needle with low tourniquet pressure
Discard the first 3-5ml and then collect into a 1 in 10 volume of trisodium citrate
Keep at room temperature, avoid shaking and test within 4 hours
Factors affecting platelet function & test results
Drugs - NSAIDS, antibiotics, antidepressants, beta-blockers, anticoagulants
Co-morbidities - renal failure, MPN, liver disease
Misc - Dextrans, radiographic contrast, expectorants
Food – fat, garlic, caffeine, turmeric, alcohol, fenugreek, onion, ginger, ginseng
Temperature, pH
Platelet count outside the range of 100-600
FGN concentration
Sample prep and handling
investigations
Platelet count:
FBC - be aware of method, impedence, light scatter, flow etc
Blood film - platelet number / clumping / size / granularity
Flow cytometry (using CD41+CD61)
Platelet Volume
Derived parameter from FBC
Platelet Aggregometry
See below
Platelet Receptor Densities
Flow cytometry for Glanzmanns (CD41,CD61), Bernard-Soulier (CD42b,CD42a)
Genetics / Genomics
Next Generation Sequencing (NGS) / High Throughput Sequencing (HTS)
Multiple genes assessed in one assay (‘Gene panel’)
Multiple techniques with varying limitations
Other Tests:
Nucleotide release assays - storage pool and release defects
Alpha Granule proteins
Electron Microscopy - abnormalities of organelles and cytoskeleton
Super resolution light microscopy
Whole Blood Aggregometry
Extended antibody panels - for rare activation disorders (Scott Syn, Stormorken Syn)
PFA-100 - no longer recommended in adult practice but see bottom of page for method
Light Transmission Aggregometry (LTA)
Method
Light passing through the sample is recorded as agonists are added to platelet-rich plasma and stirred at 37oC.
Confusingly, results may be charted against optical density or light transmission —> produces opposite curves.
Analytical Variables – see above

A few specific cases
Hydroxycarbamide – abnormal ADP and Adrenaline
A small percentage of normal population show reduced response to adrenaline
Results:
Typical graphs when plotted using light transmission:

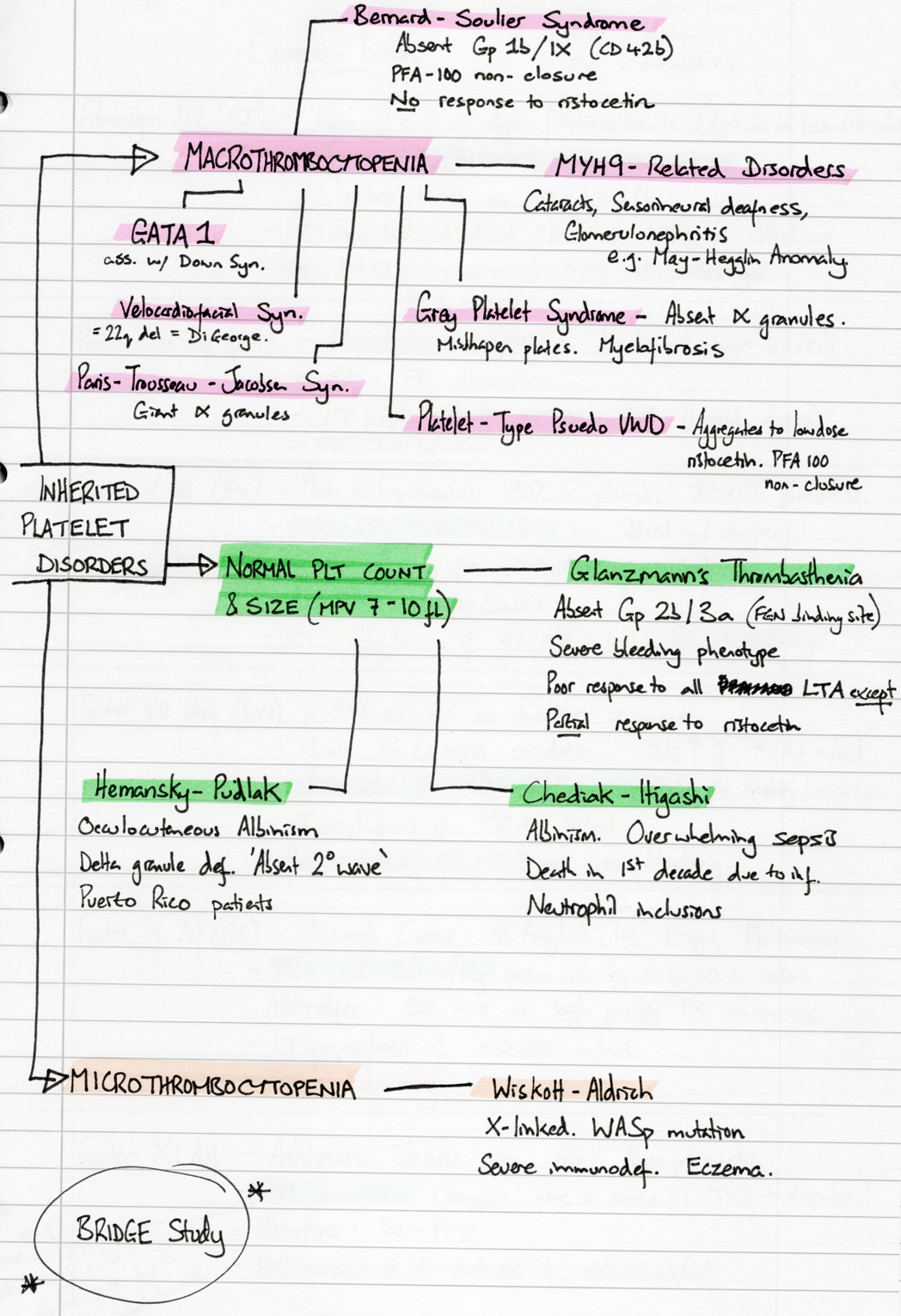
list of heritable Platelet Disorders
Glanzmann Thrombasthenia (GT)
Autosomal recessive – ITGA2B & ITGB3 gene mutations (& >100 others)
Deficiency or functional deficiency of the GpIIb/IIIa receptor
GpIIb/IIIa mediates the aggregation of activated platelets by VWF, FGN and other proteins
Presentation
Usually <5 y.o.
Purpura, epistaxis, gum bleeding (but major neonatal complications are rare)
May present later in adolescence as severe menorrhagia
Associated with angiodysplasia and GI bleeding
Natural history
Severity of bleeding diminishes with age
Except severe risk of bleeding in labour remains
Investigation
Normal platelet count & size
PFA-100 non-closure
LTA – no response except for partial aggregation with ristocetin
Flow cytometry for GpIIb and GpIIIa receptor density
Bernard-Soulier Syndrome (BSS)
Autosomal recessive (GP9, GP1BA, GP1BB) and dominant (GP1BA, GP1BB)
Deficiency or absence of GpIb/IX/V complex
GpIb/IX/V complex is a receptor for VWF —> deficiency results in defective plt adhesion
Presentation
Usually presents in childhood
Epistaxis, easy bruising, gum bleeding
Sometimes 1st diagnosed in pregnancy, or confused with ITP
Investigation
Macrothrombocytopenia (count anywhere from 30 to normal)
PFA-100 non-closure
LTA – no response to ristocetin
Flow cytometry for GpIb-alpha receptor density
Grey Platelet Syndrome
<100 cases worldwide
Storage pool disorder - Absence of alpha granules
Associated with myelofibrosis
Investigations
Macrothrombocytopenia of typical grey appearances
PFA-100 normal
Absence of alpha-granules on electron microcopy
Chediak-Higashi
Autosomal recessive – CHS gene mutation
Platelet granule abnormality
Clinical Features:
Associated with albinism
Infection + lymphoproliferative disease often results in death in 1st decade of life
Investigation
Normal platelet count
Peroxidase-positive cytoplasmic granules in neutrophils
Hermansky-Pudlak
Puerto Rican ethnicity (1 in 1800, compared to 0.5 per million worldwide)
Delta granule deficiency
Multiple genetic mutations identified, inc. HPS1, HPS4, AP3B1.
Clinical Features:
Oculocutaneous Albinsim, pulmonary fibrosis, granulomatous colitis
Early death due to fibrosis
Pattern of presenting syndrome maps to different causative gene mutation
Investigation
Normal platelet count
Absent 2o wave on LTA
Electron microscopy
myh-9 related disorders
e.g. May-Hegglin Anomaly (see photo on morphology page)
Clinical Features:
Sensorineural hearing loss, presenile cataracts, glomerulonephritis, abnormal LFTs
Wiskott-Aldrich Syndrome
X-linked, WAS mutation —> WASP protein deficiency
Clinical Features:
Severe immunodeficiency, eczema
CATCH-22 Syndrome
Deletion on chromosome —> deletes 30-50 genes, including the Gp1b gene
Multi-system disorder, CATCH only some of the features
Cleft lip
Abnormal facies
Thymus
Cardiac abnormalities
Hypocalcaemia
Even rarer syndromes
Scott Syndrome - Impaired annexin V binding.
Stormoken Syndrome - STIM1 gene. Enhanced annexin V binding. Myopathy, Hyposplenism, Hypocalcaemia
Sitosterolaemia - ABCG5, ABCG8 genes. Inability to excrete plant sterols. Atheroscelorosis, Xanthomas
Quebec Platelet Disorder - Duplication of PLAU gene. Platelet granule disorder
Management of Congenital Platelet Disorders
General Guidance
Manage at a specialist haemophilia centre with 24-hour access to care
Lifestyle
Avoid contact sports
Avoid aspirin / NSAIDs
Vaccinate for Hepatitis A & B, and monitor LFTs
Often iron deficient, replace as required
Manage pregnancy with a pre-written plan and MDT consultation.
Specifics to consider
Tranexamic Acid
Desmopressin – for plt storage pool disorders
Platelet transfusion
rFVIIa – licensed for use in Glanzmanns
Stem cell transplant
summary
Platelet Function Analyser (PFA-100)
No longer routinely used in adult practice
A measure of global platelet function
Used at the screening stage alongside PT, APTT, VWF, FVIII
A normal result may avoid the need for more difficult, time consuming tests
Method:

Results:
High negative predictive value
If PFA-100 if normal then primary haemostasis very likely to be intact
(Exceptions: Storage pool disorders, Primary secretion defects, mild type 1 VWD)
‘Non-Closure’ is typical of Glanzmann, Bernard-Soulier and Platelet-Type VWD
