Genetic haemochromatosis (Bsh 2017)
Intro
One of the most frequent genetic disorders in North Europeans
Continued absorption of iron from the small intestine despite normal/high total body iron levels leads to tissue iron deposition and subsequent organ dysfunction.
Life expectancy is normal when phlebotomy is started before cirrhosis and diabetes have developed
Normal Iron balance
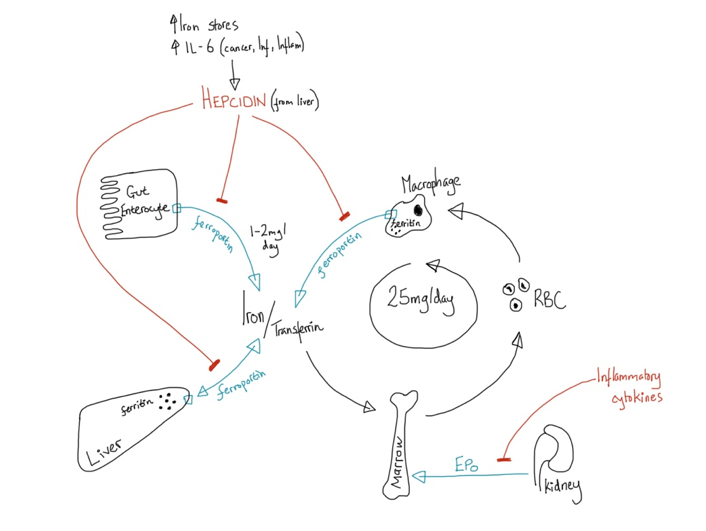
Total body iron content is around 4g, of which:
3g in haemoglobin
1g in RES macrophages (stored as ferritin)
100mg in tissue enzymes involved in respiration
4mg bound to transferrin
There is no active excretion of iron, losses occur from:
1mg/day via death of cells in the GI lumen
15-40mg/period
500mg/pregnancy
Transferrin (Tf)
Glycoprotein produced by the liver, responsible for delivering iron to cells via transferrin receptors
Tf levels are regulated by iron stores, rising in deficiency and falling in overload.
Each Tf has two iron binding domains and is capable of delivering 20-40mg of iron every 24 hours
In health, around 30% of transferrin is saturated with iron at any one time.
All cells receive iron via Tf. In addition to this, macrophages have a far greater supply via the ingestion of circulating red cells.
Iron toxicity
If serum iron levels rise --> Tf levels fall and the Tsat% rises --> non-transferrin bound iron (NTBI) starts to appear in the plasma, and is then transported into cells alongside Tf-iron.
NTBI is thought to damage cells via the Fenton reaction, free radical formation and lipid peroxidation.
HFE Gene Mutation
90% of patients with GH
Autosomal recessive inheritance of mutation in the HFE gene on chromosome 6
HFE gene codes for a glycoprotein which has complex interactions within the iron homeostasis pathways. HFE mutation results in suppression of hepcidin --> deregulation of iron efflux (see diagram above)
Two common mutations:
C282Y mutation: Cysteine --> Tyrosine at amino acid 282
H63D mutation: Aspartic acid --> Histidine at amino acid 63
(H63D mutation more common in general population, but does not usually cause iron overload)
Biochemical penetrance is variable and only 80%/50% (Male/female) of people with even the most high-risk genotype (C282Y homozygotes) will develop a raised SF/Tsat during their lifetime.
Clinical penetrance is even lower again, but from highest to lowest risk:
C282Y homozygotes
C282Y/H63D compound heterozygotes
C282Y heterozygotes
H63D homozygotes
H63D heterozygotes
(Note: It is possible that other factors (Alcohol, Metabolic Syn) play a more important role in causing hyperferritinaemia in all but the C282Y homozygotes. Some would argue that H63D mutations should not be tested for, e.g. French laboratories are not reimbursed for running this test).
Screening
Unselected population screening for the HFE gene mutation is not recommended (as only 19/16% (male/female) of people with raised SF + raised Tsat are C282Y homozygotes).
Family members of patients diagnosed with HFE GH should be offered testing for:
FBC, LFT, SF, Tsat and HFE gene mutation
Family screening is not required for family of patients with C282Y/H63D compound heterozygosity
Clinical Features
Asymptomatic
Generalised weakness and lethargy
‘Bronzed Diabetes’ – type 2 diabetes with bronze skin pigmentation
Liver fibrosis / cirrhosis
Cardiomyopathy and conduction defects
‘Painful handshake sign’ – arthropathy affecting the 2nd and 3rd metacarpophalangeal joints
Impotence
Hypogonadotrophic hypogonadism – results from severe iron loading in the pituitary
Investigation
Patients with clinical features to suggest GH should be tested for:
FBC, LFT, serum ferritin (SF) and transferrin saturation (Tsat)
HFE gene mutation should then be tested for:
All adults >30 y.o. of North European descent with unexplained SF >300/>200 and Tsat >50%/>40% (Male/Female)
C282Y Homozygotes
SF <1000, normal LFT, normal examination --> no further investigation required
SF >1000 or abnormal LFT --> refer to hepatology for fibrosis/cirrhosis investigation
(Cirrhotic patients will then typically get 6 monthly liver USS + alpha-fetoprotein levels)
Non-C282Y Homozygotes
If significant iron loading is present by MRI/liver biopsy then rare causes of iron loading should be sought
Management
All fit GH patients with biochemical iron loading:
Weekly venesection until SF 20-30 and Tsat <50% (check these monthly + weekly FBC inbetween)
Then maintain with PRN venesection, preferably as blood donation – target: normal FBC, SF <50, Tsat <50%
Homozygotes with normal iron studies, or compound heterozygotes with mild rise in iron:
Recommend regular blood donation and annual measurement of SF + Tsat
Who needs liver assessment?
Ferritin >1000 or any rise in transaminases --> refer to hepatology for fibrosis/cirrhosis assessment
Liver biopsy is no longer required for the diagnosis of HFE GH, but may still be used to assess severity of fibrosis.
Patients with liver cirrhosis are at 100-fold increased risk of primary liver cancer and require 6 monthly screening with USS and alpha-fetoprotein levels.
List of Rare Genetic Causes of iron overload
Type 2 Haemochromatosis (Juvenile Haemochromatosis)
HFE2 (syn. HJV) or HAMP gene mutations.
Causes severe iron overloading with cardiac failure and panhypopituitarism.
Type 3 Haemochromatosis (Transferrin receptor 2 deficiency)
TFR2 mutation.
European and Japanese ethnicities.
Clinical phenotype lies between Type 1 and Type 2 GH.
Type 4a Haemochromatosis (Ferroportin Disease)
Loss of function mutation in SLC40A1 (syn. FPN1) gene that encodes ferroportin
Reduced macrophage iron release --> RES iron overload.
Organ damage does not occur but patients develop iron deficiency anaemia with venesection.
Type 4b Haemochromatosis
Gain of function SCL40A1 mutation.
Similar phenotype to Type 1 GH.
Type 5 Haemochromatosis
FTH1 gene mutation.
Described in a single Japanese family.
Aceruloplasminaemia
CP gene mutation
Dystonia, ataxia, dementia (iron deposition in basal ganglia).
Hereditary Hyperferritinaemia Cataract Syndrome (HHCS)
Autosomal dominant
L-ferritin deposition in the ocular lens results in early-onset cataracts
There is no need to lower the SF level
Atransferrinaemia
TF gene mutation (Autosomal recessive)
Presents a birth with severe iron deficiency anaemia + paradoxical tissue iron overload
Benign Hyperferritinaemia
FTL gene mutation
Ferritin 400 - 6000 without tissue iron overload.
other Rare Causes of Iron Overload
African Iron Overload
Iron pots used for home brewing beer
Neonatal haemochromatosis
Related to acute liver injury, may have alloimmune element
Gaucher Disease