Non-Immune Hereditary Red Cell Membrane Disorders (icsh 2015)
Red Cell Membrane Structure

Laminated structure composed of two layers with no direct contact
Outer layer
Outer surface of phospholipid bilayer membrane. A barrier for retention of cat/an-ions
Passive outflow of K+, which is then pumped back into cells in exchange for Na+
E.g. in Hereditary Stomatocytosis the leak rate exceeds pump rate à pseudohyperkalaemia
Inner Layer
Cytoplasmic surface of the bilayer is covered by a network of spectrin and actin.
Contact points between the membrane and spectrin/actin provided by:
Band 3-Ankyrin-Protein 4.2 complex
Glycophorin 4.1 complex
Quantitative or qualitative deficiencies —> red cell deformity and shortened cell lifespan.
Diagnostic Tests
SDS-PAGE - Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Gel electrophoresis of red cell membrane proteins
Identifies and quantifies membrane contents
HS --> Reduced a/b spectrin, Ankyrin, Band 3 or Protein 4.2
HE & HPP --> Reduced protein 4.1 (and reduced a-spectrin in HPP)
Osmotic Fragility Test
14 concentrations of NaCl.
Patient and normal controls compared for concentration at which the cells lyse.
Simple but non-specific – Any fragile red cells will give a positive result
Acid Glycerol Lysis Time
Also positive for other causes of spherocytosis – AIHA, pregnancy, CKD, MDS
Osmotic Deformability Test
Plots curves of deformability that can differentiate the membrane disorders
See original guideline for sample graphs.
Eosin-5-Maleimide (EMA) Binding Test
Fluorescent test using flow cytometry
Reference ranges need to be established within each laboratory
Band 3 is the main binding site for EMA --> HS cells will show reduced binding
Results
A patient:control ratio of <0.8 consistent with HS & SAO
A ratio <0.6 seen in HPP
A normal ration is seen in HE
Confounding factors
Reticulocytes and macrocytes also show reduced binding --> equivocal results or false +
Genetic Sequencing
Not usually required where there is a family history to support diagnosis
Poor correlation between mutation and clinical diagnosis
Hereditary Spherocytosis (HS)
1 in 2000 North Europeans
Pathophysiology
75% Autosomal dominant
Defect in the Band 3-Ankyrin-Protein 4.2 complex --> Uncoupling of the cytoskeleton from the cell membrane in localized areas --> formation of spherocytes with reduced flexibility --> destroyed prematurely in spleen.
Presentation
Typically anaemia, jaundice, splenomegaly and reticulocytosis with a negative DAT.
Severity usually the same within one family, but highly variable between families.
Diagnosis
Newly diagnosed patients with a family history of HS, typical clinical features and typical lab features (spherocytes, raised MCHC and reticulocytosis) do not require further investigation.
If diagnosis is equivocal, screening test with EMA binding or cryohaemolysis is sufficiency for diagnosis.
Gel electrophoresis is the test of choice in atypical cases.
Management
Folic acid if mod-severe HS & during pregnancy
Annual Clinic Review
USS liver for gallstones every 3-5 years
Iron status
Consider Splenectomy if:
Severe disease beyond the age of 6
Moderate disease that is symptomatic and interfering with lifestyle
?Timing around cholecystectomy
If child undergoing splenectomy, cholecystectomy should be performed at the same time for symptomatic stones. Concomitant cholecystectomy for asymptomatic gallstones is controversial.
If child undergoing cholecystectomy for symptomatic gallstones, then concomitant splenectomy is controversial. It reduces the risk of bile duct stones, but exposes patient to risk of asplenic sepsis.
Remember to give alert card post splenectomy
Splenectomy Vaccinations (Green Book Jan 2020)
Men C - 2 weeks before or 2 weeks after.
Men B - 2 weeks before or 2 weeks after. Repeat after 1 month
Men ACWY - 1 month after Men C
Pneumococcal - 2 weeks before or 2 weeks after. Repeat every 5 years
Seasonal ‘flu - Annual
(Hib (Haemophilus influenza B) vaccine is no longer recommended as prevalence is so low in the UK)
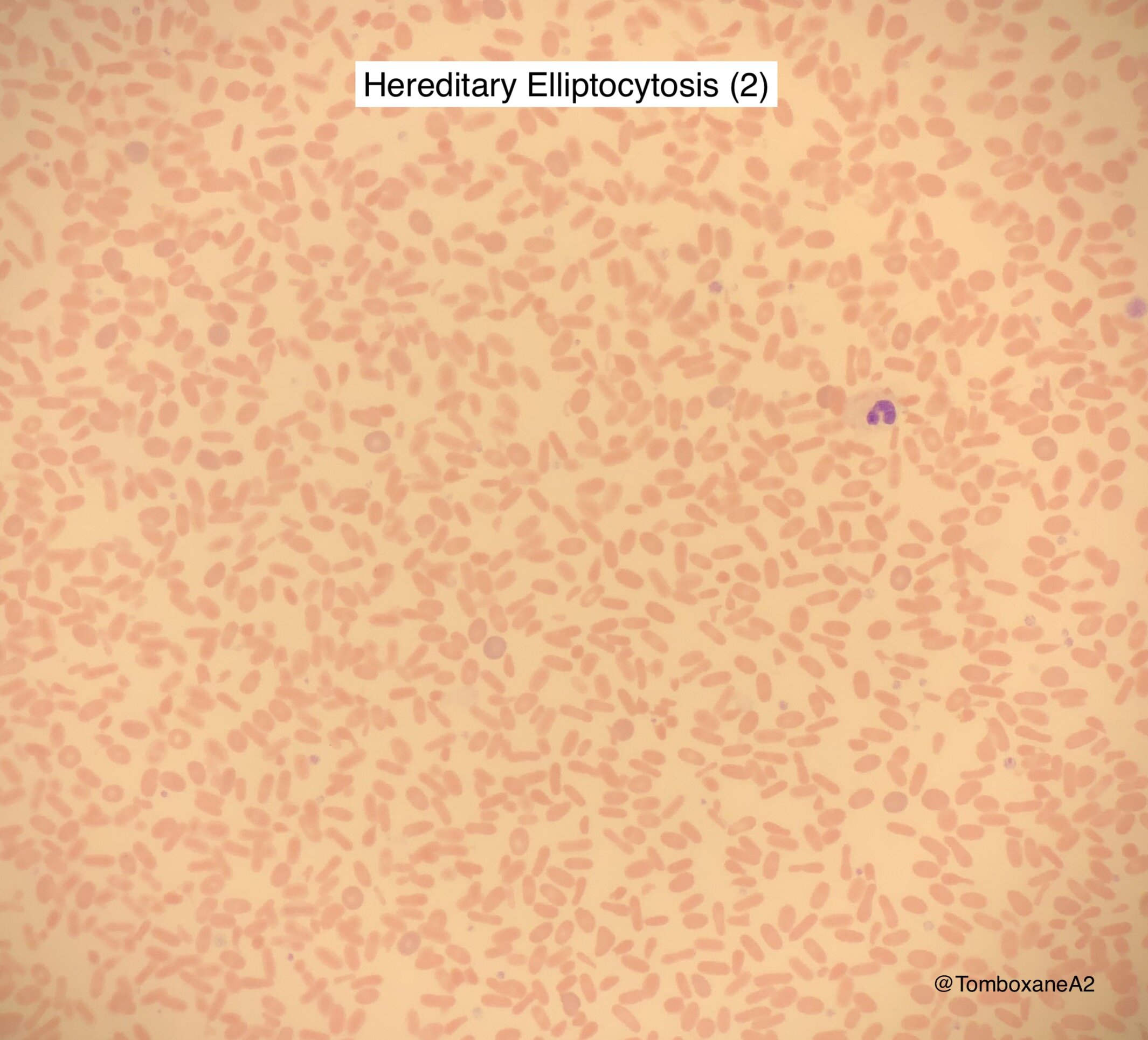
Hereditary Elliptocytosis (HE) & Pyropoikilocytosis (HPP)
Defect in the Glycophorin 4.1 complex
HE
Incidental finding in most patients with a normal Hb or mild compensated anaemia
Elliptocytes vary between 10-100% of cells seen
HPP
Rare severe form of HE causing red cell fragmentation, especially in <1 year olds.
May have requirement for transfusion in early years
Hereditary Stomatocytosis (HSt)
A group of haemolytic conditions resulting from defects in Na/K transfer
Overhydrated – moderate to severe haemolytic anaemia
Dehydrated
Cryohydrocytosis
Familial Pseudohyperkalaemia – universally asymptomatic
South East Asian Ovalcytosis (SAO)
1.5% of SE Asia population
Haemolytic in childhood and then normalizes.