Haemoglobinopathy Diagnosis (BSH 2023, uk thal standards 2016, NHS thal screening, GTG 2014)
N.B. Limited text functions on this site make this page a challenge, so all Greek characters have been swapped into the Latin.
Intro
Hbpathies are the commonest recessive monogenic disorders worldwide
Thalassaemias = Reduced production of haemoglobin E.g. a/b/d/e/g-thal
Haemoglobin variant = Altered structure of haemoglobin. E.g. HbS/D/O (b-chain), HbG (a-chain)
Thalassaemic haemoglobin variant = E.g. Hb Constant Spring (a-chain), Hb Lepore, Hb E (b-chain)
Chromosome 16 - a1, a2, z genes
Chromsome 11 - b, d, e, g genes

typical Geographic Distributions
a0 - China, Taiwan, SE Asia, Cyprus, Greece, Turkey, Sardinia
b-Thal - Anywhere other than North Europe
HbS - Africa, Greece, Southern Italy, Turkey, Middle East, India
HbE - SE Asia
HbC - West Africa
thalassaemias
alpha-Thalassamaemia
There are four alpha chain alleles (a1 and a2 genes on each copy of ch. 16). These are written as aa/aa when all four alleles are present and functioning. The number of absent or dysfunctional alleles correlates with the clinical picture as below:
-a/aa, a+ Trait
Hb normal, MCV low, MCH low. Normal electrophoresis
-a/-a, Homozygous a+ Trait
Hb normal, MCV low, MCH low (<25). Normal electrophoresis
--/aa, a0 Trait
Hb normal, MCV low, MCH low (<25). Normal electrophoresis
--/-a, HbH Disease
Hb 70-110, microcytic, hypochromic
Results in production of HbH from 4 b chains. Detectable by electrophoresis, unlike the traits.
Spectrum of clinical phenotype from asymptomatic to severe anaemia and even hydrops.
--/--, Hydrops Fetalis
Incompatible with fetal life due to absence of alpha chain synthesis.
Results in production of Hb Barts from 4 gamma chains.
aTa/aTa, Non-deletional a-thal
E.g. Saudi T
In ‘standard’ deletional a+ thal the remaining genes are upregulated to compensate.
In homozygous aTa/aTa, this does not happen --> as a result HbH disease can occur even with two functioning a genes.
b-Thalassaemia
There are two beta chain alleles (one beta gene on each copy of ch. 11).
-/b, b-Thal Trait
Hypochromic, microcytic anaemia with a raised HbA2
-/- (b0) or -/partial (b+), b-Thal Major
(Transfusion Dependent Thalassaemia (TDT) / non-dependent (NTDT) increasingly replaces major/intermedia.)
1 in 4 births where both parents are -/b
Mutations --> deletion of b gene, d&b genes or even b, d & g genes
Frequently the result of inheritance of two different mutations, i.e. compound heterozygotes.
alpha chains still produced in normal quantities and so become present in excess --> pathology of b-Thal
Clinical Features
Severe anaemia at 3-6 months when the switch from HbF to HbA occurs
Hepatosplenomegaly, Bone expansion, Iron Overload, Infection, Osteoporosis
Transfusion
?When to start – case by case decision
Vol/Freq – aim pre-Tx Hb 90-100, often equates to 2-3 units per month in adults.
d-Thalassaemia
Classified as d0 and d+, similar to b-Thal.
No clinical significance other lowering the HbA2 and so compromising the diagnosis of a co-inherited b-Thal.
d&b-Thalassameia
Classified as db0 and db+, depending on residual output of chains from the affected chromosome.
Includes Hb Lepore – a fusion mutation of the d&b genes.
egdb- Thalassaemia
Rare, results from large deletions
Severe haemolytic, hypochromic anaemia at birth. Improves after age of 3-6 months.
Only reported in heterozygotes. Homozygous state thought to be lethal at early fetal stage
Screening
Pre-conception Screening
Recommended to offer to at risk groups, i.e. most non-Northern European ethnicities (see table 1 in BSH2023)
If abnormality detected, partner should be tested
Antenatal Screening
Purpose is to detect:
1. Significant maternal hbpathy - SS, SC, S/b, HbH, b-thal intermedia
2. Maternal carrier states - AS, AC, AD, AE, AO, ALepore, b-trait, db-trait, HPFH, homozygous a0 trait
High prevalence areas = 2% or more of booking bloods are positive --> universal lab screening + FOQ
Low prevalence areas = <1% or more of booking bloods are positive --> FOQ to direct lab screening
Screening reports must be available within 3 working days
The antenatal and newborn screening programmes must be formally linked so that results can be paired and communicated between the two services with ease.

Low Prevalence Area Screening Algorithm Simplified
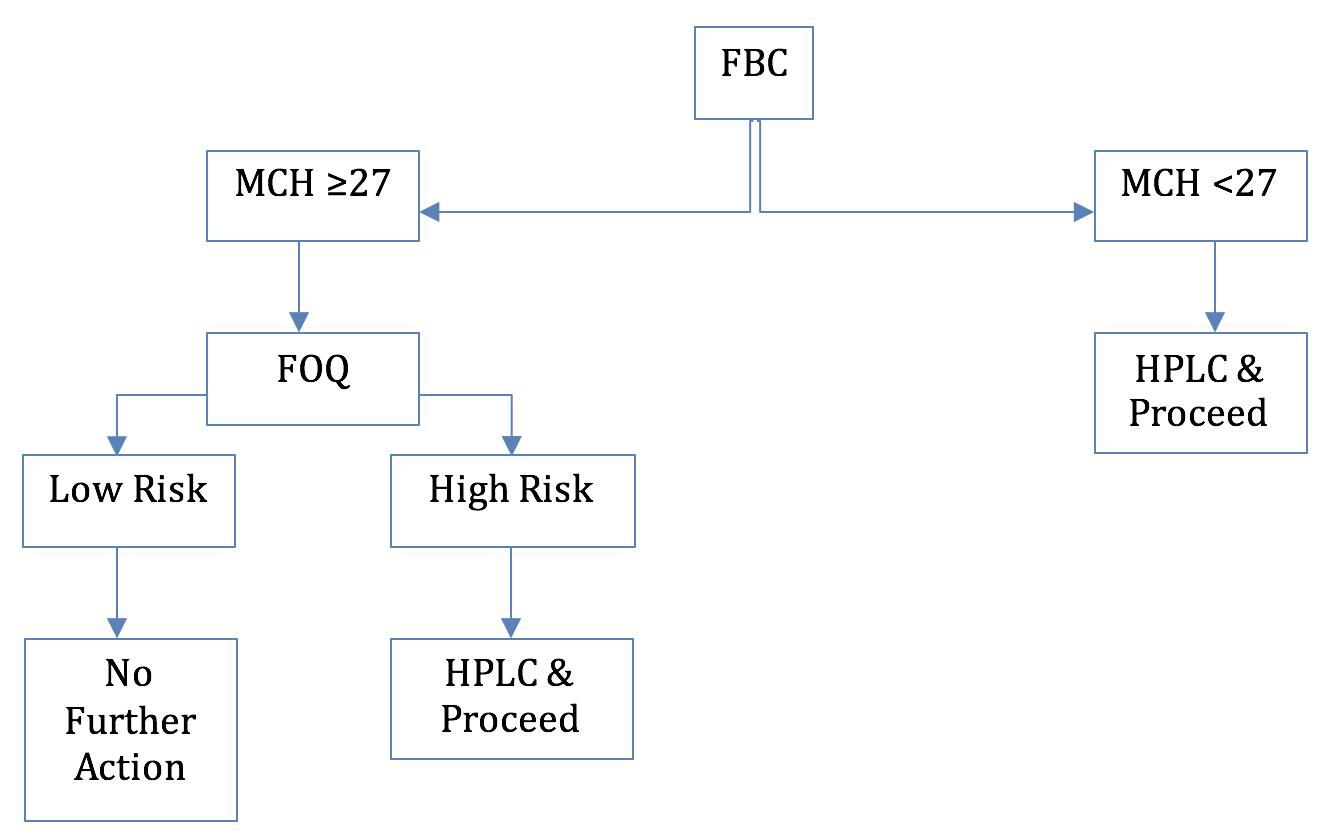
FOQ = Family Origins Questionnaire given to all pregnant women in low prevalence areas
Newborn Screening
All newborns screened for SCD (and any <1 year old arriving in the country)
Aimed at providing early diagnosis to prevent long term morbidity
May or may not detect other hbpathies
Babies with only HbF or low HbA should be followed up for further testing for b-thal major
General Testing
GP or other HCP’s can request haemoglobinopathy screening when clinically relevant and with patient’s consent
Example indications for testing:
Hydrops fetalis
Unexplained anaemia / haemolysis / splenomegaly
Unexplained microcytosis / polycythaemia / target cell poikilocytosis / anisopoikilocytosis
Unexplained cyanosis with normal oxygen sats
Pre-op Screening
Testing should be offered to all patients from high prevalence groups
A finding of SCD will inform surgical and anaesthetic management
Testing should be performed in pre-assessment clinic
If sickle solubility test used in emergency setting then must be followed up with definitive testing later
acting on an abnormal antenatal screening result
S, C, D, E, H, Lepore or O detected
Confirm finding by an alternative method, e.g. Hb electrophoresis if the initial method was HPLC
Baby’s father should be offered screening without waiting for result of confirmatory test
Raised HbA2% / HbF
Interpret as per the guides lower down this page. Test baby’s father as needed.
Isolated low MCH
DDx: Iron deficiency, heterozygous a0 (—/aa), homozygous a+ (a-/a-)
Further testing for a0 (—/aa) should be performed based on family origin. a0 usually have MCH <25.
a+ (a-/a-) is not clinically significant in itself. a+ usually have MCH 25-28
Confirm with genotyping.
Haemoglobinopathy patient cards
Hbpathy cards should be issued to patients when a major haemoglobinopathy is identified and/or when a definitive diagnosis can be made.
Hbpathy cards should not be issued for apparent alpha thal unless confirmed by DNA analysis
Testing methods
Gel/cellulose Electrophoresis
Acid/Alkaline electrophoresis infrequently use in UK now due to time restraints. Automated, high throughput methods such as HPLC and CE have taken over as workloads increase.
The test however remains simple, reliable and rapid.
If used as a screening test, confirmation should be performed with HPLC or sickle solubility testing
Useful secondary technique after HPLC
Alkaline Electrophoresis
Cellulose Acetate or Agarose Gel (pH 8.2-8.6)
Hb negatively charged and will move towards the positive anode
A split A2 band is suggestive of an a chain variant
Acid Electrophoresis
Citrate Agar or Agarose Gel (pH 6.0-6.2)
Hb complexes with agaroprotein and moves towards the positive anode
Non-complexed Hb will move toward the negative cathode.
Used to distinguish S from D, and C from E & O-Arab
Different haemoglobins form bands on the electrophoresis strip at typical locations, as shown here:

Sickle Solubility Test
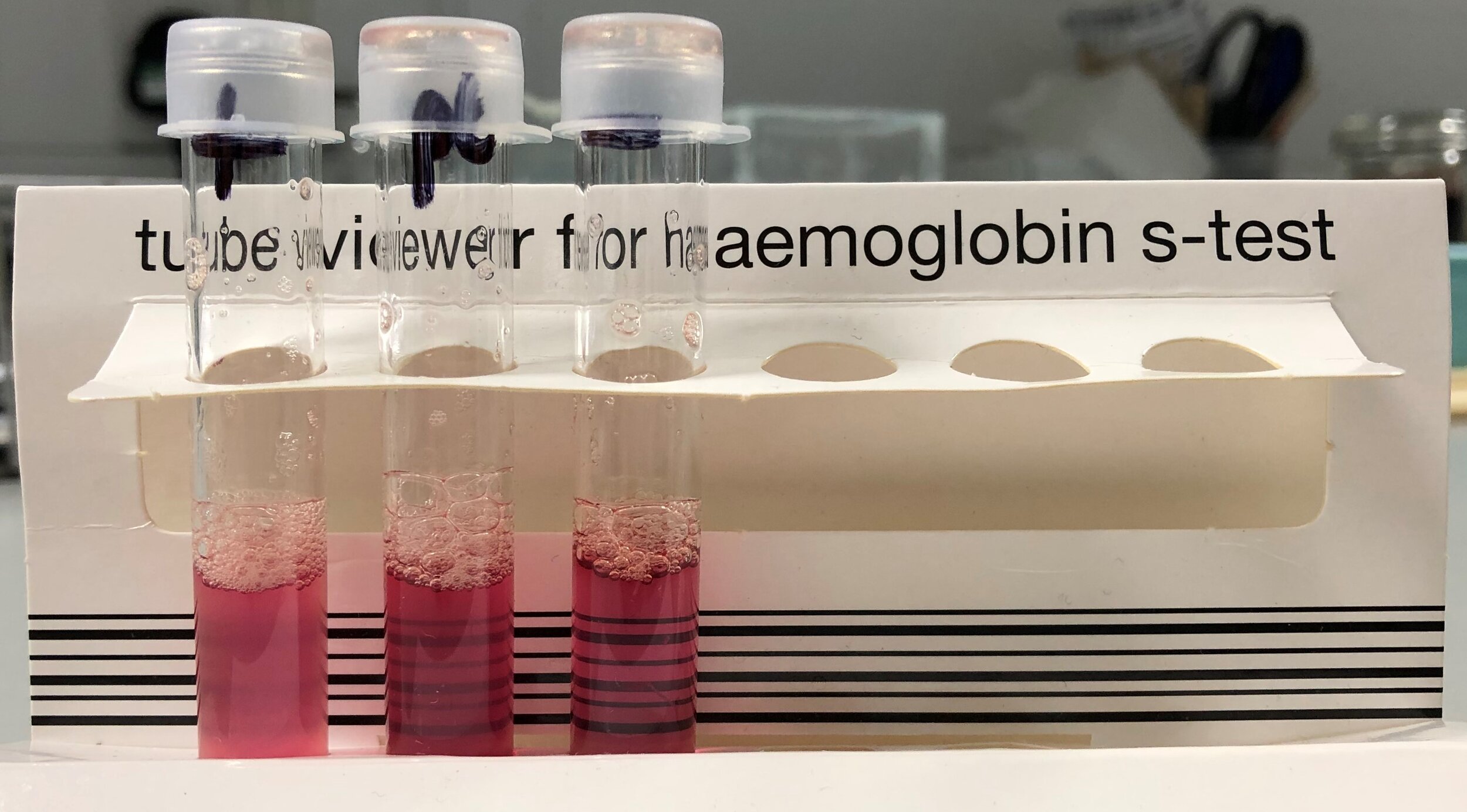
From left to right: Positive Control, Patient Sample, Negative Control
Purchased as a commercial kit
Detects HbS down to a concentration of approx. 20%
False negatives – Anaemia, Infancy, Recent Transfusion
False positives – Paraprotein, Hyperlipidaemia, Heinz Bodies, Leukocytosis
If used as a screening test, confirmation should be performed an alternative technique
HbH Preparation
HbH bodies are intracellular precipitates of 4 beta-chains.
Occur in conditions with an excess of beta chains, i.e. alpha thalassaemia
(Acquired HbH disease seen in association with MDS and MPN)
HbH precipitates with brilliant cresyl blue (a ‘supravital’ stain)
Results in golf ball cells (and Heinz bodies if hyposplenic)
Isoelectric Focusing (IEF)
Can be used to analyse whole blood, haemolysates and dried blood spots
Separates F from A or variant haemoglobins
If used as a screening test, confirmation should be performed with HPLC or sickle solubility testing
Capillary Electrophoresis (CE)
Separates A, A2, E, F, S, C, D-Punjab and G-Philadelphia
If used as a screening test, confirmation should be performed with HPLC or sickle solubility testing
Differences to HPLC
Separates E and A2
CE does not separate glycosylated fractions (?Advantageous)
Automated CE systems allow higher throughput than HPLC
high performance liquid chromatography (HPLC)
Principle of test
Mixture of molecules (normal and variant Hb) with a net positive charge are adsorbed onto a negatively charged column.
These are then eluted off in a mobile phase – a liquid containing increasing concentrations of cations flows through the column, competing for the anionic binding sites.
As the positive Hb molecules are eluted off they are detected optically and the retention time is recorded. The area under the peak can quantify each Hb.

HPLC can:
Separate A, A2, F, S, C, D-Punjab and G-Philadelphia
(E and Lepore usually co-elute with A2)
Detect previously undiagnosed diabetes through reporting of glycosylated haemoglobin.
Advantages over Haemoglobin Electrophoresis
Less labour intensive
Very small sample required
Quantification possible
Greater range of Hb’s identified
A2 detected and quantified —> easier to diagnosis b-thal
Disadvantages compared to Electrophoresis
Higher capital and reagent costs
Glycated haemoglobins
Each haemoglobin present can undergo ‘post-translational modification’, usually glycation, which affects its charge and so appears as a separate peak to the left of the original Hb.
Commonest example – the glycated A peak will increase in diabetes (HbA1c)
In SCD, there is a factitious rise in A2 as the glycated S sits in the A2 window.
interpreting HPLC Results
Low A2, 0-2%, Normal FBC
d-thal trait
d-variant (e.g. A2 prime) will produce to 2 equal peaks, in the A2 and S windows (‘Split A2’)
a-variant (e.g. G-Philidelphia) will produce to 2 unequal peaks (‘Split A2’)
Normal A2, 2-3.5%, Normal FBC
Normal.
Pitfall: Silent b-thal mutations (e.g. +1480(C—>G))can have a normal HbA2
Pitfall: Delta variants (e.g. A2 prime) give a peak in the S window. Need to add the normal and variant A2 together to get total, otherwise might miss a b-thal.
Pitfall: The HPLC trace baseline can wander up, giving an underestimated A2.
Normal A2, 2-3.5%, low MCH
a-thal trait - Indistinguishable from iron deficiency
a0 and a+ - Indistinguishable on HPLC
egdb-Thal - Once an adult, indistinguishable from a and iron def.
Therefore, if MCH <25 and a0 possible based on ethnicity —> DNA Analysis
Raised A2, >3.2% (b chain disorders)
≥3.5% + MCH <27 - Heterozygous b-thal
>4% + MCH normal - Mild b-thal, B12/folate def, liver disease, drugs, HIV
>8% - Hb Lepore heterozygous
>15% - Hb E
Pitfall: In b-thals, co-existence of a-thal will reduce A2 by 5% for each missing a gene.
E.g. HbE typically shows HbA of 30%. HbE/a+ homozygous will have 20% HbA2
Pitfall: Normal individuals with low MCH due to iron deficiency or a-thal trait, may have a HbA2 of 3.5-4% due to hyperthyroidism or drugs for HIV.
Raised HbF, >0.8%
>5% + MCH <27 - Could be heterozygous db-thal
>10% + MCH normal - Hereditary Persistance of Fetal Haemoglobin (HPFH)
HPFH
Deletional – results from db chain deletions
Non-deletional – may be a beneficial modifier
Pitfall: HPFH often associated with a co-inherited a-thal and so MCH not a reliable means to differentiate between HPFH and db-thal.
N.B. Causes of a raised HbF in infants includes:
Trisomy 13, Chronic hypoxias, Small for gestational age, HPFH
HbS
80-95% - HbSS
40% - S trait (40% not 50% as a chains prefer the normal b chains)
HbS% will rise if S trait combined with b-thal as alpha chains can no longer preferentially bind with the normal beta chains. Recently transfused patients will have a lower HbS%
Pitfall: S, C and E can carry over from previous sample and appear in the next patient’s trace.
N.B. Voxelotor
Voxelotor is a HbS polymerisation inhibitor trialled for used in sickle cell disease (NEJM 2019)
Binds to alpha globin chains —> alters structure of A, A2, F and S —> alters their positions on HPLC, CE and IEF —> double peaks / bands / difficult interpretation of results.
Extra Notes on Variants
b-variants (S, C, E, D, O) will make up roughly 50% of Hb (2 genes)
a-variants (G) will make up roughly 25% of Hb (4 genes)
If an a-variant and b-variant co-exist three peaks will form – the a, b and hybrid.
E.g.

limitations of Screening
Sensitivity/Specificty
None of the current screening techniques can identify all abnormalities
However, combining tests, e.g. HPLC + CE, has a very high sensitivity+specificity
Some cases will nevertheless require DNA analysis
Recent blood transfusion
Screening results can be misleading within 4 months of a blood transfusion
If urgent results required, consider DNA analysis
Conditions not detected by newborn screening
Some conditions are only detectable once a mature haemoglobin pattern has been established, e.g.
B-thal carriers, Hb Lepore, HPFP
Some cases of B-thal major and intermedia
Conditions not differentiated by newborn screening
Some conditions are difficult to differentiate until a mature haemoglobin pattern has been established, e.g.
HbSS versus Compound HbS + beta,delta or gamma chain thals
HbEE versus HbE/beta0 thal
Premature babies
HbA detected from approx. 30 weeks (sometimes from 24 wks)
This will prevent detection of beta chain disorders
Conditions not detected by antenatal screening
Not detected by either high or low prevalence algorithms
Silent B-thal carriers
Alpha0 (—/aa) carriers in non-high risk groups
Dominant hbpathies in biological father where mother has a negative screen
Uncommon but clinically significant hbpathies, e.g. unstable haemoglobins
Co-inheritance of B-thal and triplicated alpha in a neonate
Not detected by the low prevalence algorithm
Hb variants in northern european families
Thal carriers obscurred by B12/folate deficiency, liver disease or other causes of raised MCV/MCH
Combine b-thal + a-thal carrier
Note: Programme also not designed to detect risk of child with HbH disease.
Accurate information from family origin questionnaire
Quality of information provided affects correct use of the algorithm
Important for midwives to be well trained in guiding women through the FOQ, e.g. consider ethnic/family origin not just country of birth, consider earlier generations not just patient and partner etc.